TDP43
The Protein That Left
A single protein abandons the nucleus — and in over nine in ten people with ALS, toxic clumps of it fill dying motor neurons.
The walkthrough
Beat by beat

HOOK
0:15
01HOOK
A single protein abandons the nucleus. In the vast majority of people with ALS — well over nine in ten — toxic clumps of it fill dying motor neurons. F1
02THE NAME
The gene is TARDBP — TAR DNA-binding protein — on chromosome one, short arm. F2 Its protein, TDP-43, normally lives in the nucleus, binding thousands of RNA messages: regulating which exons are kept, which are discarded, how long each message survives. F3
03THE HUNT
TDP-43 was first found in 1995 — in HIV research, not neurons. Scientists discovered a protein that bound a key sequence in the viral genome, named it, and moved on. F4 Eleven years later, Manuela Neumann and colleagues published in Science: TDP-43 was the major protein inside the insoluble inclusions filling ALS motor neurons — present in almost every patient, confirmed the same year by an independent group. F5
04THE METHOD
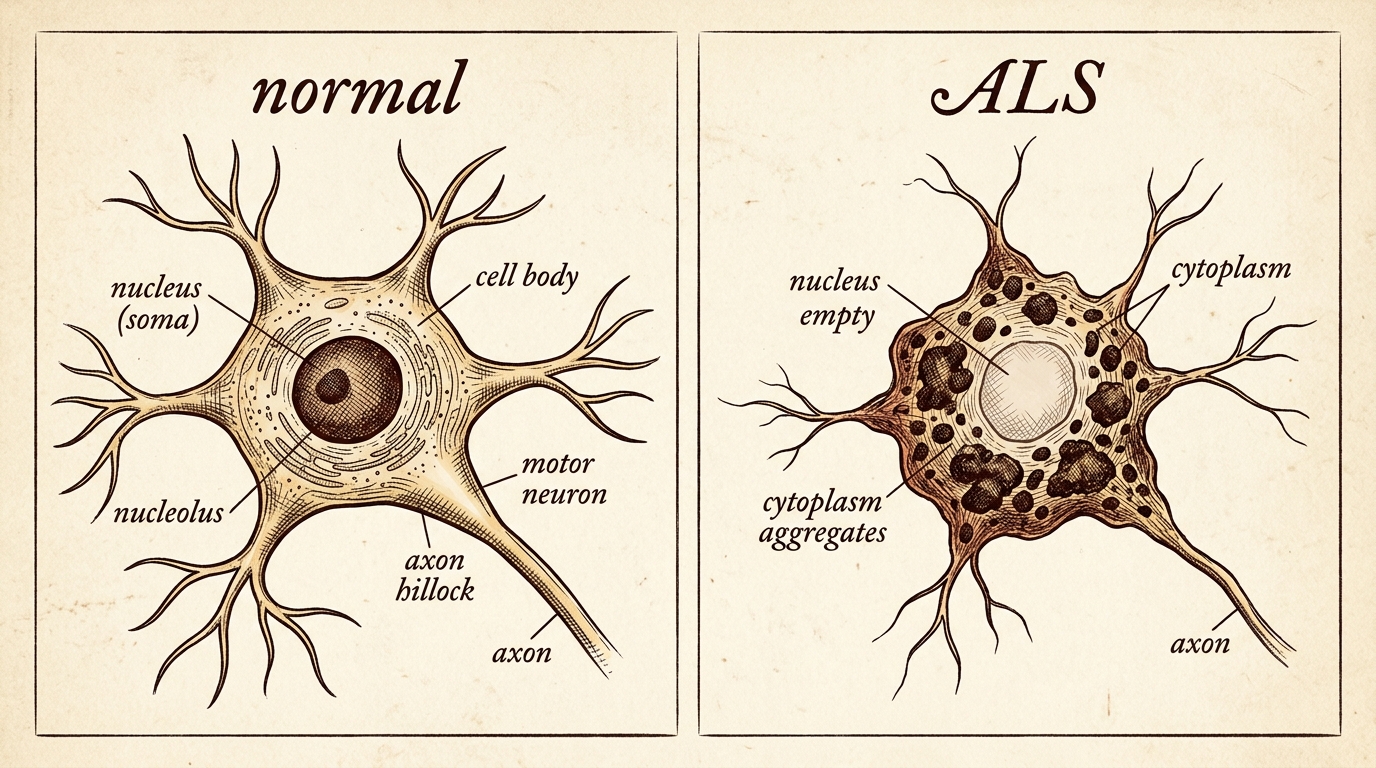
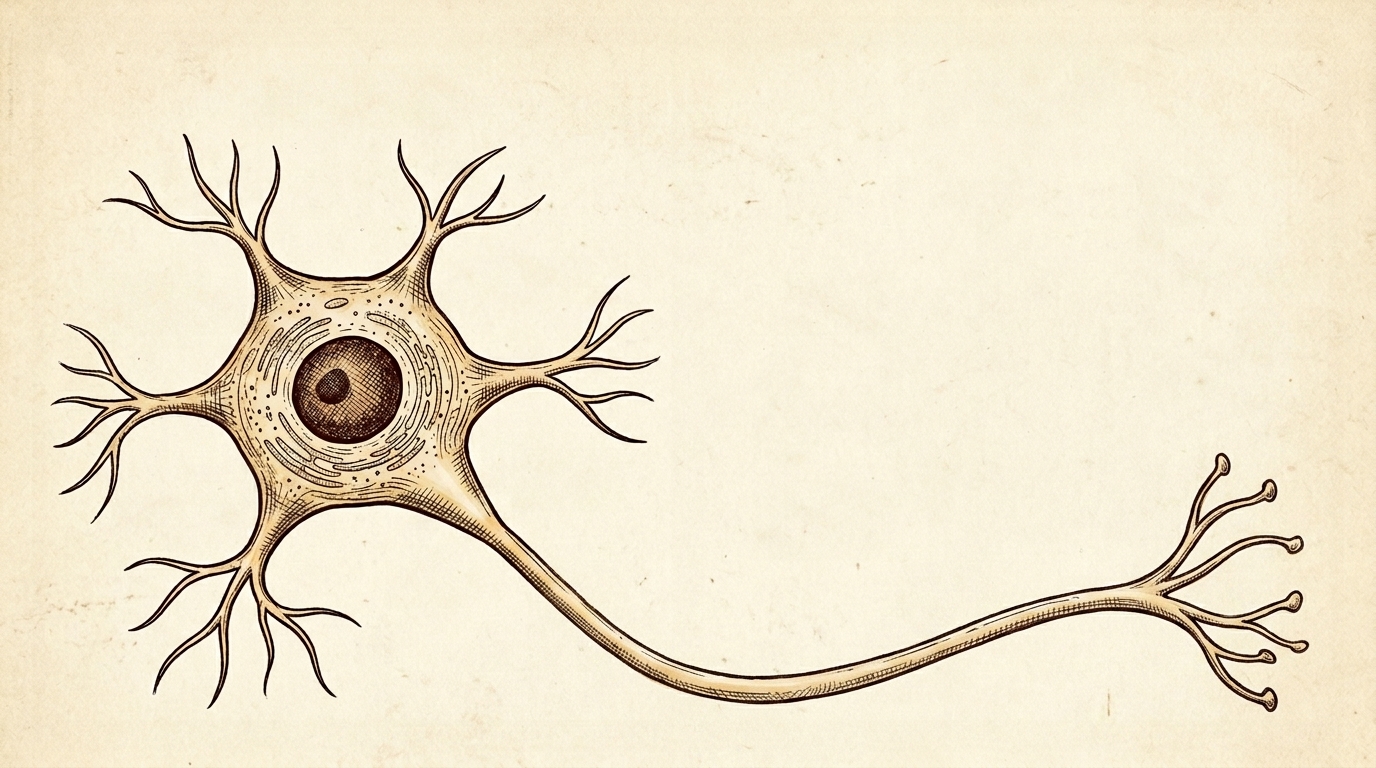
The method was immunohistochemistry. Antibodies lit up TDP-43 wherever it sat. In healthy tissue: clean, dark nuclei. In ALS tissue: the nucleus conspicuously empty, the cytoplasm full of clumps. F5
05THE MECHANISM (hero)
Normally, TDP-43 settles in the nucleus, regulating thousands of RNA messages. In disease, it is cleaved into fragments, phosphorylated, and forms insoluble aggregates in the cytoplasm. F6 A double hit: the clumps are directly toxic, and the now-empty nucleus can no longer repress hundreds of cryptic exons — hidden sequences that, when included, corrupt proteins the motor neuron depends on. F6b
06THE STAKES
ALS takes movement, speech, breathing. Most people survive two to five years from diagnosis. F7 The same TDP-43 pathology appears in nearly half of all frontotemporal dementia cases — and in LATE, a newly-named disease of ageing found in roughly one in four people over eighty-five. F7b
07THE OPEN THREAD
There is no approved disease-modifying therapy targeting TDP-43. But antisense oligonucleotides — synthetic strands that tune gene expression — are in clinical trials, targeting TDP-43 directly or its downstream effects. F8 The central challenge: TDP-43 is essential. You cannot simply remove it. The goal is to keep TDP-43 in the nucleus — or coax it back there.
08TIMELINE + SIGN-OFF
1995: discovered chasing HIV. 2006: found in every ALS case. 2019: a third disease named. The same protein. Still no cure. — The Gene Channel.
The write-up
In one line: TDP-43 is an essential nuclear protein that polices thousands of RNA messages — yet in the vast majority of ALS it deserts the nucleus and clumps in dying motor neurons, a double hit of toxic aggregates and lost RNA control that also drives much of frontotemporal dementia and the ageing disease LATE; found in 1995 while chasing HIV and pinned to ALS in 2006, it still has no approved therapy, because you cannot simply remove a protein the cell can't live without.
The gene
The gene is TARDBP — TAR DNA-binding protein — on the short arm of chromosome 1. Its protein, TDP-43, normally lives in the nucleus, binding thousands of RNA messages: regulating which exons are kept, which are discarded, and how long each message survives. It is not a bystander protein but a central piece of RNA housekeeping the motor neuron depends on.
The hunt
TDP-43 was first found in 1995 — in HIV research, not neurons. Scientists discovered a protein that bound a key sequence in the viral genome, named it, and moved on. Eleven years later, in 2006, Manuela Neumann and colleagues published in Science: TDP-43 was the major protein inside the insoluble inclusions filling ALS motor neurons, present in almost every patient — a finding confirmed the same year by an independent group (Arai et al.).
The mechanism
The method that revealed it was immunohistochemistry: antibodies lit up TDP-43 wherever it sat. In healthy tissue, clean dark nuclei; in ALS tissue, the nucleus conspicuously empty and the cytoplasm full of clumps. In disease, TDP-43 is cleaved into fragments, phosphorylated, and aggregates into insoluble inclusions in the cytoplasm. The damage is a double hit: the clumps are directly toxic, and the now-empty nucleus can no longer repress hundreds of cryptic exons — hidden sequences that, when wrongly included, corrupt proteins the neuron needs.
The stakes, and the frontier
ALS takes movement, speech, and breathing; most people survive two to five years from diagnosis. The same TDP-43 pathology appears in nearly half of all frontotemporal dementia cases, and in LATE — a newly named disease of ageing found in roughly one in four people over 85. There is still no approved disease-modifying therapy targeting TDP-43, but antisense oligonucleotides — synthetic strands that tune gene expression — are in clinical trials, aimed at TDP-43 directly or its downstream effects. The central challenge is that TDP-43 is essential: you cannot simply remove it. The goal is to keep it in the nucleus, or coax it back there.
Sources
Full claim-by-claim evidence is in references.md. Primary anchors:
- Neumann et al. (2006) Science 314:130 — PMID 17023659 — TDP-43 as the major inclusion protein in ALS/FTLD
- Arai et al. (2006) BBRC 351:602 — PMID 17084815 — independent same-year confirmation
- NCBI Gene 23435 — TARDBP — gene/locus, protein identity, RNA-binding function
- Neumann (2009) review — PMC2662455 — 1995 origin, pathology, mechanism
- Ito & Okada (2024) — PMC11651183 — cryptic-exon mechanism and therapeutic strategies
Accuracy note: The gene is TARDBP; the protein is TDP-43 — keep them distinct. TDP-43 was discovered in HIV research, not in neurons. Its pathology is not unique to ALS — the same misplaced protein underlies a large share of frontotemporal dementia and the ageing-related disease LATE. No TDP-43-targeting therapy is approved as of this writing.
The evidence
Every claim, sourced
Each [F#] you hear in the film links to the source it came from. Nothing gets narrated until every one is checked and signed off.
Sign-off
- PhD sign-off — facts above are correct; traps above stated correctly in
script.md. - Any numbers/dates verified (or narration kept qualitative).
Gate OPEN — run `gen-narration.mjs` then assets → Video.tsx → render → `writeup.md`.
- F1
The vast majority of ALS patients (well over nine in ten) have TDP-43 cytoplasmic inclusions / nuclear depletion in motor neurons
Neumann 2009 review: "TDP-43 pathology is a consistent feature in all sALS and non-SOD1-fALS" (PMC2662455 L32). Ito 2024 review says "~90% of sporadic ALS." The exception is SOD1-linked familial ALS (~2% of all ALS). TRAP: do not say "97%" without citing a specific study that uses that exact number — different reviews use 90–97%. "Well over nine in ten" or "the vast majority" is safe.
- F2
TARDBP encodes TDP-43; gene on chromosome 1 short arm (1p36.22); protein is 414 amino acids
Neumann 2009: "TDP-43 is a 414 amino acid protein encoded by the TARDBP gene on chromosome 1" (PMC2662455 L40). Locus confirmed by NCBI Gene (ID 23435).
- F3
TDP-43 normally lives in the nucleus, regulating exon splicing, mRNA stability, and RNA transport
Neumann 2009: "predominantly localized to the nucleus under normal conditions… exon skipping and splicing inhibitory activity… mRNA stabilization" (PMC2662455 L40–42). Also: Koike 2024 review of cryptic splicing / TDP-43 nuclear functions (PMC11301021).
- F4
TDP-43 first identified in 1995 in HIV research — it bound TAR DNA in the HIV-1 LTR
Ou SH et al., "Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs." J Virol. 1995;69(6):3584–96. PMID 7745700. Referenced by Neumann 2009 (PMC2662455 L40–41) as ref [55]: "It was first cloned as a human protein capable of binding to the transactive response DNA of human immunodeficiency virus type 1."
- F5
In 2006, Manuela Neumann and colleagues (Science) identified TDP-43 as the major protein in ALS ubiquitinated inclusions; confirmed independently the same year by a Japanese group
Two landmark simultaneous papers: (1) Neumann M, Sampathu DM, …, Trojanowski JQ, Lee VM. "Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis." Science 2006;314:130–133. PMID 17023659. (2) Arai T, Hasegawa M, …, Oda M. "TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis." Biochem Biophys Res Commun 2006;351:602–611. PMID 17084815. Both cited in PMC2662455 as refs [5] and [6]. The discovery method was monoclonal antibody screening + 2D SDS-PAGE + LC-MS, then confirmed by immunohistochemistry — the narration simplifies this to IHC, which is accurate as confirmation step.
- F6
In disease, TDP-43 is cleaved into fragments, phosphorylated, ubiquitinated, and forms insoluble cytoplasmic aggregates
Neumann 2009: "disease-specific bands at ~25 kDa, ~45 kDa… due to N-terminal truncation, hyperphosphorylation and ubiquitination of TDP-43 in FTLD-U and ALS" (PMC2662455 L14). The ~25 kDa and ~35 kDa C-terminal fragments are the aggregation-prone species.
- F6b
Nuclear TDP-43 loss allows cryptic exons to be included in STMN2 and UNC13A mRNAs, corrupting proteins motor neurons depend on
Ito 2024: "Nuclear TDP-43 loss in patients with ALS results in incorporation of a premature polyA tail in STMN2 and inclusion of a cryptic exon in the UNC13A transcript, leading to reduced function" (PMC11651183 L40). Koike 2024 review specifically covers this mechanism (PMC11301021). Original cryptic exon papers: Ling JP et al., Science 2015;349:650 (PMID 26251643); Klim JR et al., Nat Neurosci 2019;22:167 (PMID 30643218).
- F7
ALS: most people survive two to five years from diagnosis
Paulukonis et al. 2015 (PMC4501568): "median survival time post-diagnosis was 2.6 years"; range cited in introduction: "20 to 48 months" (~1.7–4 years from onset). The "2–5 years" range is the standard clinical figure and is accurate.
- F7b
TDP-43 pathology in ~half of FTD cases; LATE affects roughly one in four people over 85
Neumann 2009: FTLD-TDP (formerly FTLD-U) is the most common FTLD subtype, ~45–50% of cases. LATE prevalence: Wolk et al. 2025 (PMC11772733 L39): "≈25–40% of those ≥85 years of age." The original LATE naming paper is Nelson PT et al., Brain 2019;142:1503–1527 (PMID 31039256 / doi:10.1093/brain/awz099). "Roughly one in four over eighty-five" is the conservative end of the 25–40% range — acceptable.
- F8
ASOs are in clinical trials targeting TDP-43 directly or its downstream effects (STMN2, UNC13A). No approved disease-modifying therapy yet.
Ito 2024 (PMC11651183): tofersen (SOD1 — FDA accelerated approval); QRL-201 (STMN2 splice-switching, ANQUR study NCT05633459); BIIB105 (ATXN2 — discontinued May 2024). Keuss et al. 2024 (PMC11230273): UNC13A splice-switching ASOs restore synaptic function. TRAP: "no approved disease-modifying therapy targeting TDP-43" is correct — tofersen is approved for SOD1-ALS only, not TDP-43 proteinopathy.