HBB
One Letter
One misplaced letter — a T where there should be an A — bends a round red cell into a sickle. It took seventy-five years to undo.
The walkthrough
Beat by beat



HOOK
0:26
01HOOK
In your DNA, a single letter is out of place — one T where there should be an A `F1`. That one swap, in one gene, bends a round blood cell into a brittle crescent `F2`. And for the seventy-five years after we first understood it, there was no cure `F3`. This is the story of that gene.
02THE NAME
The gene is called HBB. It sits near the tip of chromosome 11 `F4`, and its only job is to build beta-globin — one of the four protein chains in hemoglobin, the molecule that carries oxygen in your blood `F5`. Two alpha chains, two beta. Get one letter wrong in the beta, and everything downstream changes.
03THE HUNT I
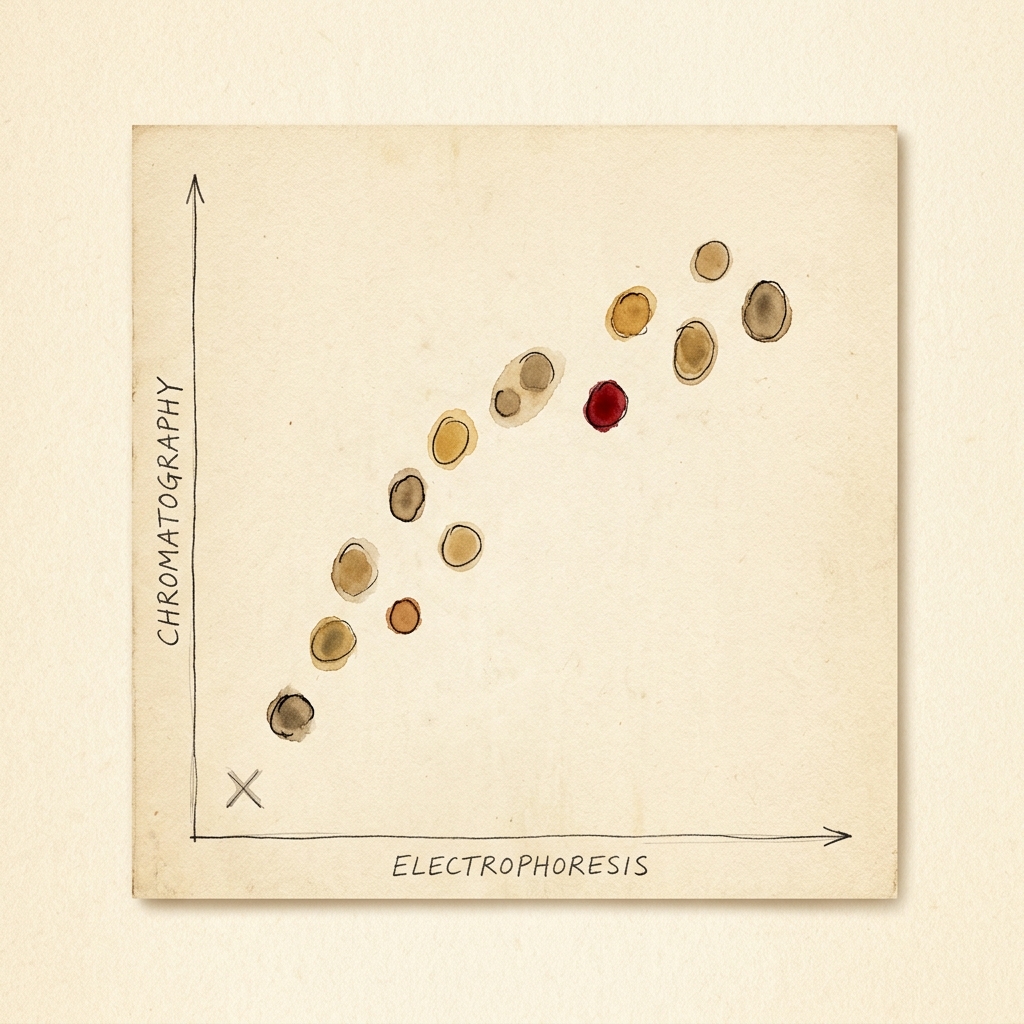
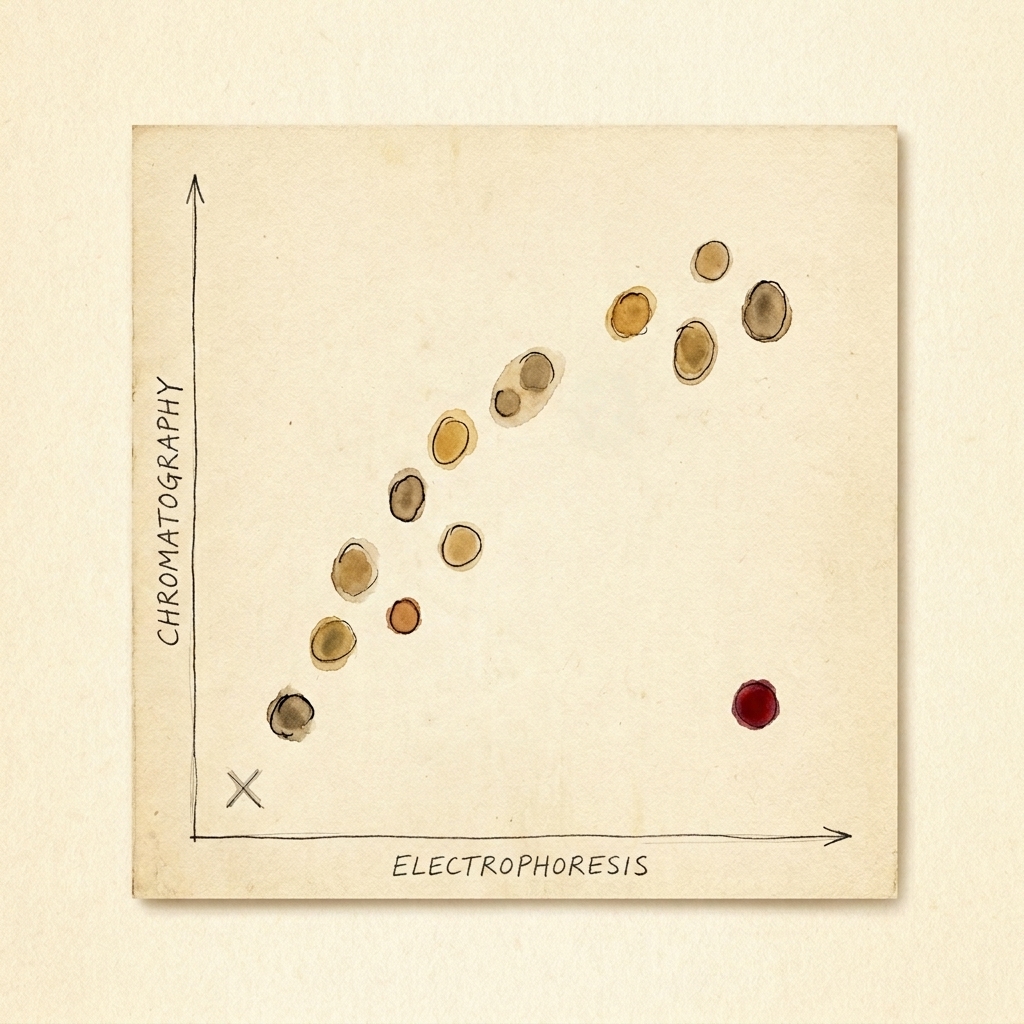
In 1949, Linus Pauling and his team did something no one had done before. With a technique called electrophoresis, they showed that the hemoglobin of a sickle-cell patient carried a different electric charge than healthy hemoglobin `F6`. The flaw wasn't in the cell — it was in a single molecule. They called it the first "molecular disease." `F7`
04THE HUNT II
But which part of the molecule? In 1956 and '57, Vernon Ingram pinned it down to a single amino acid: the sixth building block of the beta chain — a glutamic acid — was swapped for a valine `F8`. One amino acid, out of the 146 in beta-globin `F8b`. It was the first time a disease was traced to one change in one protein `F9`.
05THE MECHANISM (hero)
Here is why one swap matters. The gene is read in three-letter words. The healthy codon G-A-G spells glutamic acid; flip the middle letter and G-T-G spells valine `F10`. Glutamic acid loves water; valine hides from it. So when oxygen runs low, the mutant hemoglobin molecules lock together into stiff fibers `F11`, stretching the round cell into a sickle. Those sickled cells snag in blood vessels — and that is the pain, and the damage `F12`.
06THE STAKES
For decades that was the whole, cruel story: understand the disease almost perfectly, and still be able to do almost nothing. Sickle cell remains one of the most common inherited diseases in the world `F13` — millions of people living in the gap between knowing and curing.
07THE OPEN THREAD
Then, in December 2023, the gap closed `F14`. A therapy called Casgevy became the first approved medicine built on CRISPR gene-editing `F15`. And here's the twist: it doesn't repair the broken HBB gene at all. Instead it switches a different gene back on — reawakening the fetal hemoglobin you made before birth, which doesn't sickle `F16`. Seventy-five years after Pauling, a single infusion can free a patient from the crises `F17`.
08TIMELINE + SIGN-OFF
One gene. One letter. Seventy-five years from a charge on a gel to a cure in a needle. The biology is solved — the open question now is who can reach a cure that costs millions `F18`. — The Gene Channel.
The write-up
In one line: A single A→T substitution in the HBB gene turns one amino acid of beta-globin from glutamic acid into valine — and that lone change is enough to bend a round red blood cell into a sickle. It took 75 years to go from understanding that to curing it.
The gene
HBB sits near the tip of the short arm of chromosome 11 (band 11p15.4) and codes for beta-globin, one of the four protein chains of adult hemoglobin (two alpha, two beta — Hb A = α₂β₂), the molecule that ferries oxygen around your body. Beta-globin is just 146 amino acids long. Change one of them, and the oxygen-carrier you depend on can turn against you.
The hunt (1949 → 1957)
In 1949, Linus Pauling, Harvey Itano, S. J. Singer and Ibert Wells ran hemoglobin from healthy people and from sickle-cell patients through an electrophoresis cell and watched the two migrate differently: the sickle hemoglobin carried a slightly different electric charge. The defect, they argued, wasn't in the cell or the tissue — it was in a single kind of molecule. They named the category that paper created: the first "molecular disease." Crucially, they had not yet found what was different about the molecule.
That came from Vernon Ingram. In 1956–57, using protein "fingerprinting" (digesting the chain into peptides and separating them), he located the difference to a single spot: the sixth amino acid of the beta chain, normally glutamic acid, was replaced by valine. One substitution, in one protein — the first time a human disease was pinned to a change that precise. (Modern HGVS nomenclature writes the variant as HBB c.20A>T, p.Glu7Val, counting the initiator methionine; the classic literature — and this video — use the mature-protein numbering, position 6.)
The mechanism
The gene is read three letters at a time. The healthy codon GAG specifies glutamic acid; flip the middle base — A→T — and it becomes GTG, which specifies valine. Glutamic acid is hydrophilic; valine is hydrophobic. That swapped residue creates a sticky patch on the surface of the hemoglobin molecule, and when oxygen is low, the mutant molecules (HbS) polymerize into stiff fibers. Those fibers distort the normally round, flexible red cell into a rigid sickle, which lodges in small blood vessels — the cause of the vaso-occlusive crises, pain, and cumulative organ damage of sickle cell disease.
The 75-year gap, and the cure (2023)
For most of the twentieth century we understood sickle cell almost perfectly and could do very little about it — it remains one of the most common inherited diseases in the world. Then, on December 8, 2023, the FDA approved Casgevy (exagamglogene autotemcel), the first approved medicine built on CRISPR gene-editing.
The elegant twist: Casgevy does not repair the HBB mutation at all. Instead it uses CRISPR-Cas9 to disable the erythroid-specific enhancer of the BCL11A gene in a patient's own blood stem cells. BCL11A is the switch that, after infancy, shuts off fetal hemoglobin (HbF, α₂γ₂). Knock the switch out, and the patient's cells start making fetal hemoglobin again — and because HbF has no beta chains, it cannot sickle. The edited cells are infused back, and in trials the recurrent pain crises largely disappeared. One molecule understood in 1949; one infusion, 75 years later.
The open thread isn't the biology anymore — it's access. A one-time list price around $2.2M, plus the hospital course it requires, puts this cure out of reach for most of the people who carry the gene. That gap is the next story.
Sources
Full claim-by-claim evidence is in references.md. Primary/authoritative anchors:
- Pauling L, Itano HA, Singer SJ, Wells IC. "Sickle Cell Anemia, a Molecular Disease." Science 110:543 (1949). https://www.science.org/doi/10.1126/science.110.2865.543
- Ingram VM. (1956, 1957) single amino-acid substitution in sickle hemoglobin — see https://www.pnas.org/doi/10.1073/pnas.0406677101
- HBB gene (location 11p15.4, structure): NCBI Gene/GTR 3043 — https://www.ncbi.nlm.nih.gov/gtr/genes/3043/ ; MedlinePlus — https://medlineplus.gov/genetics/gene/hbb/
- HbS variant HBB c.20A>T (p.Glu7Val), rs334: ClinVar — https://www.ncbi.nlm.nih.gov/clinvar/RCV000016573.11/
- Casgevy mechanism (BCL11A enhancer → HbF) + Dec 8 2023 approval: PMC10913280 — https://pmc.ncbi.nlm.nih.gov/articles/PMC10913280/ ; Drugs.com — https://www.drugs.com/history/casgevy.html
Accuracy note: the discovery beats are deliberately kept distinct — Pauling (1949) showed sickle cell is a molecular disease; Ingram (1956–57) identified the amino-acid substitution. And Casgevy edits BCL11A to reactivate fetal hemoglobin; it does not correct HBB.
The evidence
Every claim, sourced
Each [F#] you hear in the film links to the source it came from. Nothing gets narrated until every one is checked and signed off.
Sign-off
- PhD sign-off — facts correct; the two ⚠️ traps stated correctly in
script.md. (Signed off 2026-06-09.) - F18 — narration kept qualitative ("costs millions"); ~$2.2M U.S. list price noted here for the write-up.
- Locus 11p15.4 (current NCBI/GRCh38) accepted. Position-6 classic framing (Glu6Val) accepted for narration.
Gate OPEN → narration + render may proceed.
- F1
HbS = a single base substitution A→T in HBB (codon 6, c.20A>T, rs334)
ClinVar lists the Hemoglobin S variant as NM_000518.5(HBB):c.20A>T; rs334 single A>T base change
- F2
The mutation makes round red cells deform into sickles
Sickle cell anemia mechanism
- F3
~75 years from first understanding (1949) to a cure (2023), with no cure in between
Arc spans Pauling 1949 → Casgevy 2023 (F6, F14)
- F4
HBB sits near the tip of chromosome 11's short arm — band 11p15.4
NCBI Gene/GTR: HBB (Gene ID 3043) cytogenetic location 11p15.4, GRCh38 NC_000011.10. (Note: older sources say 11p15.5.)
- F5
Beta-globin is one of 4 chains of adult hemoglobin (2 α + 2 β); hemoglobin carries oxygen
"In adults, hemoglobin typically consists of four protein subunits: two subunits of beta-globin and two subunits of alpha-globin."
- F6
1949, Pauling et al.: electrophoresis showed sickle vs normal hemoglobin differ in electric charge
Pauling, Itano, Singer & Wells, Science 110:543 (Nov 25 1949); HbS ~3 more positive charges by free-boundary electrophoresis
- F7
They coined the first "molecular disease"
Title + thesis of the 1949 paper
- F8⚠ commonly confused
Ingram (1956–57), not Pauling, found the single amino-acid swap: Glu→Val at position 6 of the β-chain
Ingram, Nature 1956 & 1957: a single amino-acid difference, Glu6→Val. HGVS: p.Glu7Val (counts initiator Met); legacy: Glu6Val/E6V
- F8b
β-globin is 146 amino acids long
Mature human β-globin chain length
- F9
First disease traced to a single amino-acid change in a single protein ("father of molecular medicine")
Ingram's 1957 result described as the first such demonstration
- F10
Codon GAG (Glu) → GTG (Val); the middle base A→T
rs334: GAG→GTG at codon 6, single A>T → E6V
- F11
When deoxygenated, mutant HbS polymerizes into stiff fibers
Valine creates a hydrophobic contact → deoxy-HbS polymerization
- F12
Sickled cells lodge in vessels → pain + organ damage (vaso-occlusion)
Vaso-occlusive crises
- F13
Sickle cell is one of the most common inherited diseases (~100k in US)
"afflicts approximately 100,000 people in the U.S."
- F14
December 2023: a cure arrived
Casgevy FDA approval Dec 8 2023
- F15
Casgevy = first approved medicine using CRISPR gene-editing
"first medicine available in the United States to treat a genetic disease using the CRISPR gene-editing technique"
- F16⚠ commonly confused
It does NOT fix HBB — it edits the BCL11A erythroid enhancer to reactivate fetal hemoglobin (HbF, α2γ2), which lacks β-chains and doesn't sickle
"used to edit … at the erythroid-specific enhancer region of the BCL11A gene, which [de-represses] fetal hemoglobin (HbF)"
- F17
A single infusion can free patients from vaso-occlusive crises
Trial: eliminated recurrent VOCs in the large majority of treated patients; one-time ex-vivo edited autologous infusion
- F18
The cure costs millions; access is limited
Casgevy U.S. list price ~$2.2M — verify exact figure before final narration
- —
(timeline) HBB sequenced in the 1970s
Marotta, Wilson, Forget & Weissman, J Biol Chem 1977 (β-globin cDNA sequence)