CFTR
The Salt and the Gate
A child whose skin tasted of salt was once a death sentence. The story of a broken gate in the cell membrane — and the drugs that reopen it.
The walkthrough
Beat by beat


HOOK
0:39
01HOOK
There is an old European folk saying: woe to the child whose brow tastes of salt when kissed — for he is bewitched, and soon must die `F1`. For centuries no one knew why a baby's skin could taste of salt — only that it foretold an early death. That salt was the first clue to one of the most common fatal inherited diseases in people of European descent `F2` `F3`. This is the story of the gene behind it.
02THE NAME
The gene is CFTR, on chromosome seven `F4`. Its job is to build a tiny gate in the surface of your cells — a channel that lets chloride, one half of ordinary salt, pass through `F5`. Where chloride goes, water follows; and that thin film of water is what keeps the mucus in your lungs and gut loose and flowing `F6`. In your sweat glands, the very same gate pulls salt back into the body before sweat reaches the skin `F7`. Break the gate, and the salt stays behind — on the brow of a newborn.
03THE HUNT
For most of history, no one could point to the broken gate. Then, in nineteen eighty-nine, three teams — led by Lap-Chee Tsui, with Francis Collins and John Riordan — ran it down `F8`.
04THE METHOD
And here is what made it a landmark: they had no idea what the protein was, or what it did. They hunted the gene purely by its address on chromosome seven — walking, and then jumping, along the DNA until they reached it `F9`. It was one of the first times a disease gene was caught by its position alone, before anyone knew its function.
05THE MECHANISM (hero)
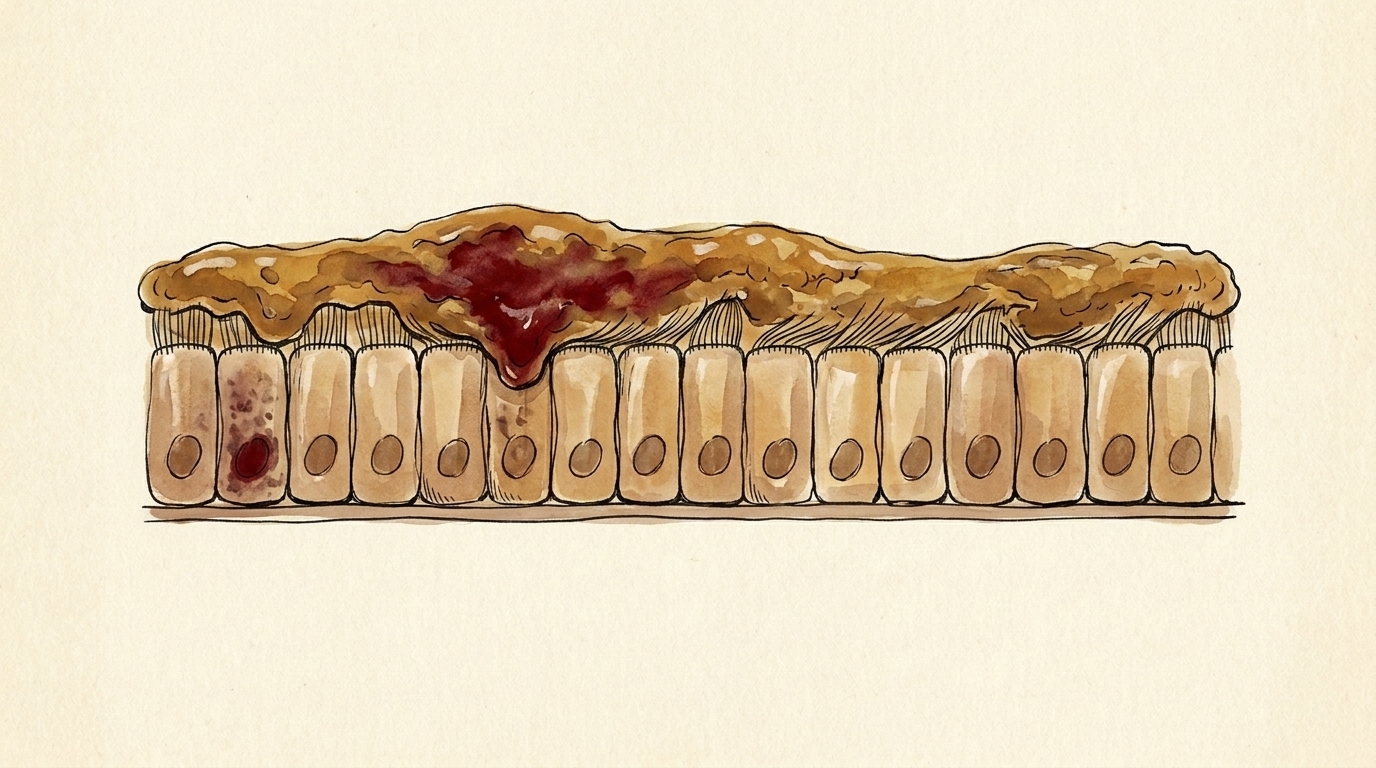
Now, the most common mutation. It is not a swapped letter — it is a deletion. Three letters of DNA, gone, erasing a single building block of the protein: a phenylalanine, at position five-oh-eight `F10`. The reading frame stays intact — almost the entire channel is still there `F11`. But missing that one piece, it folds wrong. And your cells run a quality-control line that destroys misfolded proteins — so the channel is scrapped before it ever reaches the surface `F12`. No gate, no chloride, no water — and the mucus turns thick and sticky, clogging the lungs and breeding infection `F13`.
06THE STAKES
For a long time, that was a sentence of its own. A child born with cystic fibrosis a few decades ago often did not live to start school `F14`. Understanding the gene, by itself, changed almost nothing.
07THE OPEN THREAD
Then came an unlikely kind of fix — not a gene edit, but a pill. In twenty-twelve, the first of these drugs, ivacaftor, learned to prop a faulty gate open `F15`. And in twenty-nineteen, a triple combination called Trikafta went further: two of its molecules coax the misshapen channel to fold correctly and slip past quality control to the surface, while the third holds the gate open `F16`. For the roughly ninety percent of patients who carry that deletion, it reshaped the disease — though it is not a cure, it must be taken for life, and it still cannot help the patients whose mutations make no protein at all `F17`. A child born with CF today is expected to live into their sixties `F18`.
08TIMELINE + SIGN-OFF
One gene, found by its address. A missing piece the cell mistakes for trash. Thirty years from the gene to a pill that helps the gate fold right. The biology is solved — the open question now is who can reach the medicine, and what becomes of the patients whose genes make nothing to rescue. — The Gene Channel.
The write-up
In one line: CFTR builds a chloride gate in the cell surface; the most common mutation deletes a single building block so the gate folds wrong and the cell destroys it before it ever arrives — and the modern fix isn't a gene edit but a pill that coaxes the gate to fold right and stay open.
The gene
CFTR sits on the long arm of chromosome 7 (7q31.2) [F4]. It builds a channel in the surface of your
cells that lets chloride — one half of ordinary table salt — pass through [F5]. Where chloride goes, water
follows, and that thin film of water is what keeps the mucus in your lungs and gut loose and flowing [F6]. The
same channel runs in two directions depending on the tissue: in the airway it secretes chloride to hydrate
mucus, while in the sweat duct it reabsorbs chloride back into the body before sweat reaches the skin [F7].
Break the gate and the salt stays behind — which is why, for centuries, a baby whose brow tasted of salt was
said to be bewitched and doomed [F1]. That salty skin is the molecular signature of one of the most common
fatal inherited diseases in people of European descent, and the basis of the diagnostic sweat test [F2] [F3].
The hunt
For most of history no one could point to the broken gate. In 1989, three teams — led by Lap-Chee Tsui
in Toronto, with Francis Collins and John Riordan — ran it down [F8]. What made it a landmark was the
method: they had no idea what the protein was or what it did. They hunted the gene purely by its address on
chromosome 7 — chromosome walking and jumping — until they reached it [F9]. It was one of the first times a
disease gene was caught by its position alone, before anyone knew its function (positional cloning).
The mechanism
The most common mutation is F508del — and it is not a swapped letter, it is a deletion. Three DNA letters
are gone, erasing a single amino acid: a phenylalanine at position 508 [F10]. Crucially, the deletion is
in-frame — the reading frame stays intact and almost the entire channel is still built [F11]. But missing
that one piece, it folds wrong, and the cell's quality-control machinery destroys misfolded proteins — so the
channel is scrapped in the endoplasmic reticulum before it ever reaches the surface [F12]. No gate, no chloride,
no water — and the mucus turns thick and sticky, plugging the airways and breeding infection [F13]. A few
decades ago, a child born with cystic fibrosis often did not live to start school, and simply understanding the
gene changed almost nothing [F14].
The stakes, and the frontier
The turn came from an unlikely direction — not a gene edit, but a pill. In 2012, ivacaftor (a
potentiator) became the first drug to treat the underlying defect, propping a faulty gate open [F15]. In
2019, the triple combination Trikafta went further: two corrector molecules coax the misshapen channel
to fold correctly and slip past quality control to the surface, while the potentiator holds the gate open
[F16]. For the roughly 90% of patients who carry at least one F508del, it reshaped the disease — but it is
not a cure: it must be taken for life, and it does nothing for patients whose mutations make no protein at
all for the drugs to act on [F17]. A child born with CF today is now expected to live into their sixties
[F18]. The biology is solved; the open question is who can reach the medicine — and what becomes of the
patients whose genes make nothing to rescue.
Sources
Full claim-by-claim evidence (with the commonly-confused points flagged) is in references.md.
Primary anchors:
- Gene / protein: NCBI Gene 1080 (CFTR) · UniProt P13569 · MedlinePlus: CFTR
- Discovery (1989): Science 245 — Rommens et al. (walking & jumping), Riordan et al., Kerem et al.
- F508del: ClinVar 7105 — c.1521_1523del (p.Phe508del), in-frame · CFTR2.org · PMC4101494 (F508del misfolding)
- Therapies: Vertex — Kalydeco/ivacaftor (2012) · FDA — Trikafta (2019)
- Disease / outlook: StatPearls: Cystic Fibrosis (NBK493206) · CF Foundation — life expectancy
Accuracy note: This episode is built on points that are easy to get wrong, and states each carefully —
(1) the sweat-duct direction (CFTR reabsorbs chloride in the duct; it secretes in the airway — failure
gives salty skin and dehydrated airway mucus, by opposite net direction in each tissue) [F7]; (2) F508del is
an in-frame deletion of one residue (Phe508; Ile507 is retained) — not a frameshift and not "two codons"
[F10] [F11]; (3) discovery credit is three-way (Tsui as lead; Collins's chromosome-jumping technology) and it
was one of the first positional-cloning successes, not literally the first [F8] [F9]; (4) modulators are
not a cure or gene therapy, are lifelong, and do nothing for no-protein (Class I) mutations [F17]; (5)
survival "into the sixties" is the median predicted survival for babies born today, not the current average
age at death [F18].
The evidence
Every claim, sourced
Each [F#] you hear in the film links to the source it came from. Nothing gets narrated until every one is checked and signed off.
Sign-off
- PhD sign-off — facts above are correct; the ⚠️ traps are stated correctly in
script.md. (Signed off 2026-06-10.) - Numbers kept qualitative where they drift yearly (survival "into their sixties" ← ~66, 2025 registry; "~90%" coverage).
**Gate OPEN** → narration + render may proceed.
- F1
Old European folk saying: a child whose brow tastes of salt when kissed is bewitched / will die young
Widely cited proverb: "Woe to the child who tastes salty from a kiss on the brow, for he is cursed and soon will die." ⚠️ Provenance is soft — quote the proverb, call it "an old European folk saying"; do not assert a specific year/country as fact.
- F2
Salty sweat/skin is the hallmark of CF (the molecular signature; basis of the diagnostic sweat test)
Sweat chloride test is the diagnostic gold standard: ≥60 mmol/L diagnostic, 30–59 intermediate, <30 unlikely
- F3
One of the most common fatal inherited diseases in people of European descent
"the most common lethal genetic disorder in populations of Northern European descent"; carrier ~1 in 25; incidence ~1 in 2,500–3,500 white newborns. ⚠️ Keep the ancestry qualifier — CF is much rarer (not absent) in other populations.
- F4
The gene is CFTR, on chromosome 7 (7q31.2)
NCBI Gene (CFTR, Gene ID 1080): location 7q31.2, GRCh38 NC_000007.14
- F5
CFTR builds a cell-surface channel that lets chloride through (chloride = half of NaCl)
ABC-superfamily protein that "functions as a chloride channel"; UniProt: "Mediates the transport of chloride ions across the cell membrane" (cAMP/PKA-regulated; also permeable to HCO₃⁻). ⚠️ Structurally ABC-family but functions as a gated channel, not a classic pump.
- F6
Chloride transport drives water movement → keeps mucus thin and flowing
"The transport of chloride ions helps control the movement of water in tissues, which is necessary for the production of thin, freely flowing mucus."
- F7⚠ commonly confused
In the sweat duct, CFTR normally reabsorbs chloride back into the body; when it fails, salt stays in sweat → salty skin
⚠️ Direction trap: duct = CFTR reabsorbs Cl⁻ (failure → salty skin); airway = CFTR secretes Cl⁻ (failure → dehydrated mucus). Same channel, opposite net direction by tissue. Do not say "secretes into sweat."
- F8
1989: gene identified by three teams — Lap-Chee Tsui (lead, Toronto/SickKids) with Francis Collins and John Riordan
Three back-to-back papers, Science 245, 8 Sep 1989 (Rommens; Riordan; Kerem). ⚠️ Credit is three-way (Tsui lead, Collins = chromosome-jumping tech); first authors ≠ PIs.
- F9
Method: found by positional cloning — chromosome walking and jumping — by position alone, before the protein/function was known; a landmark
Paper title: "Identification of the Cystic Fibrosis Gene: Chromosome Walking and Jumping." Routinely cited as a landmark of positional cloning / reverse genetics. ⚠️ "one of the first," not literally first (DMD 1986 preceded).
- F10⚠ commonly confused
Most common mutation = F508del: a deletion of three DNA letters removing one amino acid — a phenylalanine — at position 508
ClinVar Var. 7105:
NM_000492.3(CFTR):c.1521_1523del (p.Phe508del)— "in-frame CTT deletion … in-frame deletion of a phenylalanine at codon 508." Legacy ΔF508. ⚠️ One residue lost (Phe508), Ile507 retained — the 508-vs-507 ambiguity is naming convention only; never say "two codons." ~85–90% of patients carry ≥1 copy. - F11⚠ commonly confused
The deletion is in-frame (no frameshift) — almost the entire channel is still made
c.1521_1523delCTT removes one codon; "preserves the integrity of the reading frame" (ClinVar 7105). The near-complete protein is why a corrector can rescue it. ⚠️ Do not imply a frameshift/truncation.
- F12
F508del misfolds → destroyed by the cell's quality-control machinery (ER-associated degradation) → little/no channel reaches the surface (Class II defect)
"The F508del CFTR, instead of functioning as a regulated chloride channel on the cell surface, is retained in the endoplasmic reticulum (ER) and subject to proteasome-mediated degradation." (+ secondary gating defect; rescued protein retains partial function)
- F13
No surface channel → no chloride/water → thick, sticky mucus clogs lungs (and ducts) and breeds infection
"Thickened mucus secretions … result in mucous plugging with obstruction … an environment optimal for bacterial growth is created within the airways."
- F14
Historically, a child with CF "often did not live to start school" (≈ death before age 5)
Pre-modern-care: "most children with the disease didn't survive past age 5."
- F15
2012: ivacaftor (Kalydeco) — first CFTR modulator, a potentiator that holds the channel gate open
FDA approval Jan 31, 2012; "first medicine to treat the underlying cause"; potentiator "opens them to allow chloride ions to move"; initial indication ages 6+ with a G551D gating mutation. ⚠️ Don't merge with the Trikafta indication.
- F16
2019: Trikafta (elexacaftor/tezacaftor/ivacaftor) — triple combo: two correctors (help it fold/traffic to the surface) + the potentiator (holds the gate open)
FDA approval Oct 21, 2019; "first triple combination therapy … for the most common cystic fibrosis mutation"; two correctors + one potentiator.
- F17⚠ commonly confused
~90% of patients carry ≥1 F508del; not a cure / not gene therapy; taken for life; does nothing for no-protein (Class I/nonsense) mutations
Indication: CF patients aged 2+ with ≥1 F508del or another responsive CFTR mutation — "estimated to represent ~90% of the CF population." ⚠️ Modulators "do not respond … because there is no protein for the modulators to act on" for Class I. (Age expanded from 12+ at launch to 2+.)
- F18
A child born with CF today is expected to "live into their sixties"
Median predicted survival ≈ 66 yrs (born 2021–2025, 2025 CF Foundation Registry). ⚠️ This is median predicted survival for those born now — not current average age at death, and lower for modulator-ineligible patients. Narration kept qualitative ("into their sixties").