C9ORF72
The Stutter
A short stretch of DNA, stuttered hundreds of times over — the most common genetic cause of ALS and frontotemporal dementia.
The walkthrough
Beat by beat

HOOK
0:25
01HOOK
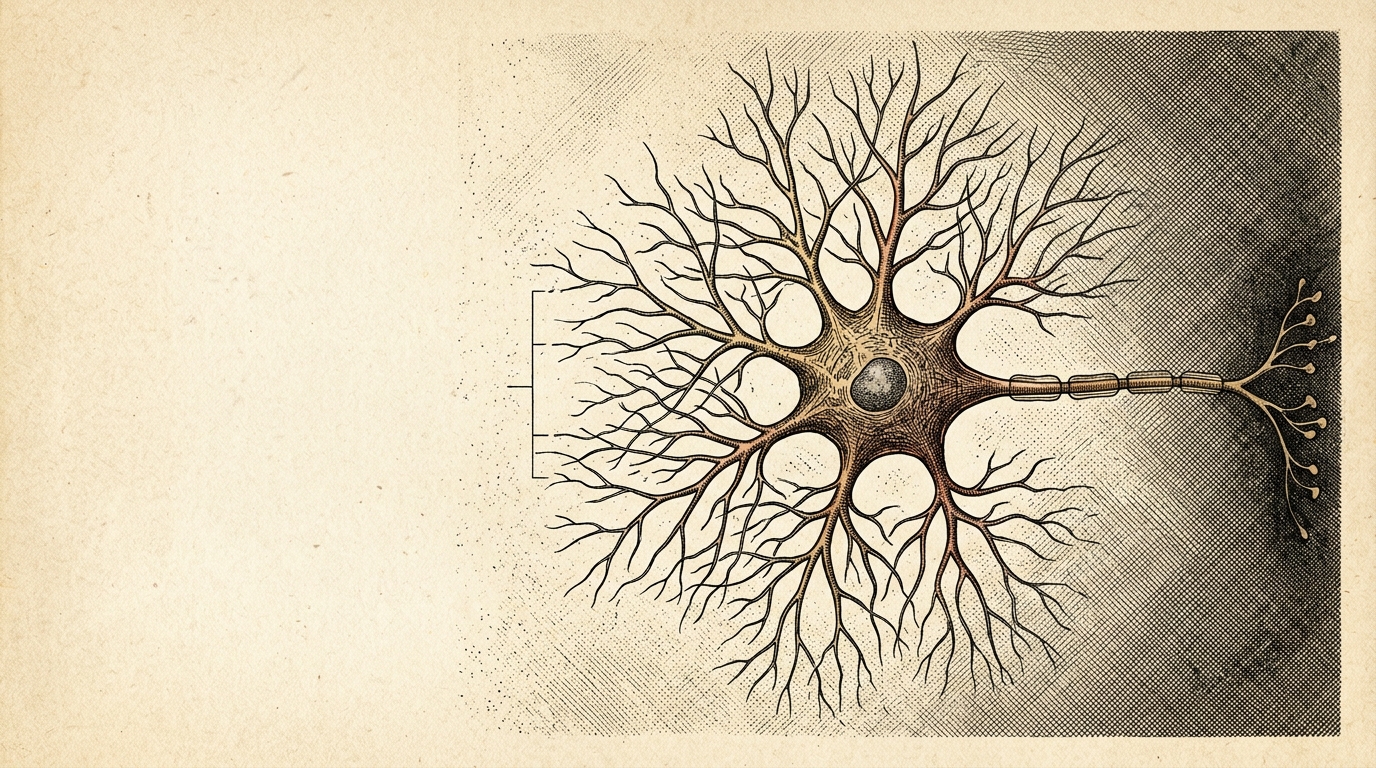
A single genetic stutter — six letters, repeated hundreds of times — is the most common known genetic cause of both ALS and frontotemporal dementia `F1`. Two different brain diseases. One mutation. This is C9ORF72.
02THE NAME
The gene is C9ORF72 — chromosome nine, open reading frame seventy-two. It sits on the short arm of chromosome nine, at band nine-p-twenty-one-point-two `F2`. Its protein helps cells break down and recycle worn-out components — a process called autophagy `F3`. But C9ORF72 is not famous for what it makes. It's famous for a six-letter sequence buried in its first intron — and what happens when that sequence loses count.
03THE HUNT
By 2011, geneticists tracing the chromosome-nine link in families with ALS and frontotemporal dementia had hit a wall — the sequence looked completely normal. Rosa Rademakers at the Mayo Clinic had spent years chasing it alongside her colleague Mariely DeJesus-Hernandez `F4` `F10`. On a different continent, a team that included Alan Renton was closing in from a separate angle. Neither group knew how close the other was. They coordinated. Both published on the same day, in the same journal, in the same issue `F5`. Not a broken letter. A short passage that would not stop repeating.
04THE METHOD
Both teams published on the same day — October twentieth, two thousand and eleven — in the same journal `F5`. Using a technique called repeat-primed PCR, they found the same thing: a six-letter sequence — G-G-G-G-C-C — repeated in the first intron of C9ORF72. In healthy people, it repeats two to ten times `F6`. In patients with ALS or frontotemporal dementia, it was repeating hundreds of times. Some patients carried thousands `F6`.
05THE MECHANISM (hero)
What makes C9ORF72 uniquely cruel is that this expansion poisons the cell three ways at once `F7`. First, it partially silences the gene — less C9ORF72 protein means the cell's recycling machinery runs slow. Second, the expanded repeat RNA folds into G-quadruplex structures that pool in the nucleus as molecular foci, sequestering the RNA-binding proteins the cell needs to manage its own transcripts. Third, that same repetitive sequence gets read in both directions without a conventional start signal — producing a swarm of sticky proteins called dipeptide repeats. The arginine-rich ones are especially toxic to neurons. Motor neurons die, or frontal-lobe neurons die, depending on which population gives way first. Same mutation. Different victims.
06THE STAKES
C9ORF72 is the most common known genetic cause of ALS — roughly forty percent of familial cases. And of frontotemporal dementia — roughly twenty-five percent `F8`. For families where the expansion runs, the question is not whether the stutter is there. It's when it will speak.
07THE OPEN THREAD
The leading strategy has been to silence the repeat — antisense oligonucleotides engineered to destroy the repeat-containing RNA before it can cause damage `F9`. Early trials reached their molecular targets: the toxic transcripts fell in spinal fluid. But in patients, the disease didn't slow. The mechanism runs three ways, and quieting one has not been enough. Newer approaches target the toxic proteins directly, or aim to restore autophagy. No treatment yet changes the course.
08TIMELINE + SIGN-OFF
Two thousand and eleven — the stutter found, simultaneously, by two teams. Two thousand and thirteen — the toxic proteins identified. Two thousand and twenty-two — first large trials completed. One stutter. Three poisons. Two diseases. The question now is whether we can reach all three arms at once. — The Gene Channel.
The write-up
In one line: A six-letter sequence that repeats hundreds of times in one gene causes the most common inherited form of both ALS and frontotemporal dementia — and a three-pronged mechanism that has so far defeated every attempt to treat it.
The gene
C9ORF72 (chromosome 9 open reading frame 72, Gene ID 203228) sits at 9p21.2 on the short arm of chromosome 9. Its protein contains a DENN domain that acts as a guanosine exchange factor (GEF) for a family of small GTPases called Rabs, and it forms a complex (with SMCR8 and WDR41) that helps recruit the ULK1 autophagy initiation complex. In plain language: C9ORF72 helps cells run their internal recycling system. When the protein is absent or reduced, autophagy slows and damaged components accumulate.
But the gene is not famous for its protein. It is famous for what sits in its first intron: a hexanucleotide repeat sequence, GGGGCC, whose copy number varies enormously between individuals.
The mutation
In healthy people, the GGGGCC repeat is present 2–10 times (the large majority of the population has fewer than 10 copies; up to ~23 copies has been observed without pathology). More than 30 copies is considered pathogenic. In patients with ALS or frontotemporal dementia who carry the expansion, the repeat typically runs to 700–1600 copies or more — sometimes several thousand. Detecting these expansions requires a specific PCR method (repeat-primed PCR) designed to catch sequences that loop back on themselves; standard sequencing misses them entirely, which is why the mutation went undetected for so long.
The hunt
By the late 2000s, geneticists had mapped a major ALS/FTD susceptibility locus to chromosome 9p21 through linkage studies of large affected families, but fine-mapping found nothing — no mutations, no obvious structural variants. Two independent teams spent years chasing it. On October 20, 2011, both published simultaneously in Neuron:
- DeJesus-Hernandez et al. (Mayo Clinic), Neuron 72(2):245–256 (PMID 21944778; PMC3202986)
- Renton et al. (an international consortium), Neuron 72(2):257–268 (PMID 21944779; PMC3200438)
Both found the same GGGGCC repeat expansion in C9ORF72. It was, at the time, the largest genetic contribution to a common adult-onset neurodegenerative disease ever identified in a single variant.
The mechanism
What makes C9ORF72 distinctive — and therapeutically difficult — is that the expansion does not act through one mechanism. It acts through three, simultaneously:
-
Loss of function. The expansion partially suppresses C9ORF72 transcription, reducing the protein's expression. Impaired autophagy means damaged proteins and organelles clear more slowly from neurons.
-
RNA foci. The expanded repeat RNA folds into stable G-quadruplex structures and accumulates in the nucleus as dense foci. These foci sequester RNA-binding proteins (RBPs) that the cell needs to splice and export its transcripts, causing widespread downstream disruption of RNA metabolism.
-
Dipeptide-repeat (DPR) proteins via RAN translation. The repeat is translated in both sense and antisense directions without a conventional AUG start codon — a process called repeat-associated non-ATG (RAN) translation, first described for C9ORF72 in 2013 (Ash et al., Science PMID 23959777; Mori et al., Science PMID 23393558). Five DPR species are produced: poly-GA, poly-GR, poly-PR, poly-PA, and poly-GP. The arginine-rich ones — poly-GR and poly-PR — are especially toxic, disrupting nucleocytoplasmic transport, phase-separated condensates, and mitochondrial function.
Which brain regions are most affected determines the clinical presentation: if motor neurons of the spinal cord and brainstem fail first, the patient develops ALS; if neurons of the frontal and temporal cortex fail first, they develop FTD; many patients develop features of both (ALS-FTD). The same expansion, different downstream targets — which is unusual even among neurodegeneration genes.
The stakes
C9ORF72 is the most common known genetic cause of ALS (~40% of familial ALS, ~8% of all ALS) and of frontotemporal dementia (~25% of familial FTD, ~6% of sporadic FTD). It is, in frequency, the dominant genetic driver of two of the most prevalent and devastating adult-onset neurodegenerative conditions.
For carriers identified before symptoms, the question is purely one of time: penetrance is high but not complete, and age of onset varies widely (typically 40s–70s). There is currently no way to predict which disease a carrier will develop, or when.
The frontier
The therapeutic approach that has advanced furthest is RNA targeting: antisense oligonucleotides (ASOs) engineered to bind the repeat-containing transcripts and trigger their degradation. Two programmes reached human trials:
-
BIIB078 (Biogen / Ionis Pharmaceuticals): an ASO targeting the sense-strand transcript. Phase 1 study in ALS patients (2022): well-tolerated; reduced dipeptide-repeat proteins in CSF. But clinical measures showed no benefit — and at higher doses, a trend toward faster decline. Development discontinued.
-
WVE-004 (Wave Life Sciences): an ASO targeting the aberrant repeat-containing transcript. FOCUS-C9 Phase 1b/2a: achieved 48–50% reduction in poly(GP) DPR in CSF (a clean biomarker hit). But again, no clinical benefit on ALSFRS-R functional scale. Discontinued.
These failures illustrate the central challenge: the mechanism runs three ways. Silencing one arm of a three-pronged disease has not been sufficient. Next-generation strategies under investigation include approaches that simultaneously address loss of function and gain-of-function toxicity, direct targeting of the DPR proteins (particularly poly-GR and poly-PR), restoration of nucleocytoplasmic transport, and combinatorial ASO strategies.
No treatment yet changes the course of C9ORF72-associated ALS or FTD.
Sources
Full claim-by-claim evidence is in references.md. Primary anchors:
- DeJesus-Hernandez et al. 2011 — PMC3202986
- Renton et al. 2011 — PMID 21944779
- GeneReviews: C9ORF72 — NBK268647
- C9ORF72 protein / autophagy — PMC6829620
- Ash et al. 2013 (DPR proteins) — PMID 23959777
- Mori et al. 2013 (RAN translation) — PMID 23393558
- NCBI Gene C9ORF72 (203228)
Accuracy note: Two ⚠️ traps in this episode. (1) The normal repeat range: narration says "2 to 10" — conservative and correct for the vast majority; healthy individuals can carry up to ~23 repeats, and the precise pathogenic threshold (~30+) is noted in references.md. (2) The ASO trial failures: BIIB078 and WVE-004 were Phase 1 / Phase 1b trials, not powered for definitive efficacy — the failure is informative but the interpretation requires care. Narration states only "the disease didn't slow," which is accurate.
The evidence
Every claim, sourced
Each [F#] you hear in the film links to the source it came from. Nothing gets narrated until every one is checked and signed off.
Sign-off
- PhD sign-off — facts correct; the F6 ⚠️ (normal repeat range — narration uses conservative "2 to 10"), F7 ⚠️ (three mechanisms, relative contributions debated — narration lists all three without ranking), and F9 ⚠️ (ASO trial failures — phase 1/1b, not full efficacy trials) all stated correctly.
- Confirm "2013" for DPR timeline beat in signoff (Ash et al. PMID 23959777 and Mori et al. PMID 23393558 — both Science 2013).
Gate OPEN when both boxes are checked → run `gen-narration.mjs`. Then assets → Video.tsx → render → `writeup.md`.
- F1
The C9ORF72 repeat expansion is the most common known genetic cause of BOTH ALS (~40% familial) AND frontotemporal dementia (~25% familial)
GeneReviews NBK268647 confirms this for both conditions; most common genetic cause of each
- F2
C9ORF72 chromosomal location: 9p21.2
NCBI Gene ID 203228, cytogenetic location 9p21.2, GRCh38 coords 27,546,546–27,573,866
- F3
C9ORF72 protein is involved in autophagy — helps cells recycle worn-out components
C9ORF72 contains a DENN domain, acts as a GEF for Rab GTPases, recruits the ULK1 complex to the phagophore, regulates autophagosome formation and lysosomal fusion
- F4
Two independent teams discovered the expansion in 2011 after years of chromosome-9 linkage work
DeJesus-Hernandez (Mayo Clinic) and Renton (international consortium), both linked to chr9p21 ALS-FTD families
- F5
Both papers appeared in Neuron on October 20, 2011
DeJesus-Hernandez et al., Neuron 72(2):245-256, Oct 20 2011 (PMID 21944778); Renton et al., Neuron 72(2):257-268, Oct 20 2011 (PMID 21944779) — published same day, same issue
- F6⚠ commonly confused
Normal: 2–10 GGGGCC repeats (up to ~23 in healthy individuals; >30 considered pathogenic). Disease: hundreds to thousands — typical expansions 700–1600+, sometimes more
DeJesus-Hernandez 2011 established threshold; 93.7% of healthy people ≤10 repeats; 99.9% ≤20 repeats. Narration says "2 to 10" (conservative/round) — acceptable since most healthy individuals are in this range; the gray zone (20–30) and the ≤23 tail are noted here.
- F7⚠ commonly confused
The expansion acts through THREE simultaneous mechanisms: (1) haploinsufficiency / partial silencing of C9ORF72 → autophagy impaired; (2) repeat RNA forms G-quadruplex structures → nuclear foci sequester RNA-binding proteins; (3) RAN (repeat-associated non-ATG) translation in both directions produces dipeptide-repeat proteins (DPRs); arginine-rich DPRs (poly-GR, poly-PR) are especially toxic to neurons
All three mechanisms are well-established. ⚠️ Trap: the loss-of-function vs gain-of-function debate is ongoing; the relative contribution of each mechanism is still debated. The narration correctly states all three without claiming we know which is primary.
- F8
C9ORF72 accounts for ~40% of familial ALS and ~25% of familial FTD
GeneReviews: ~40% familial ALS (range 30–50% across populations; 46% in Finnish cohort); ~25% familial FTD (range varies by geography)
- F9⚠ commonly confused
ASO trials reached molecular targets (toxic transcripts/DPRs fell in CSF) but showed no clinical benefit — both discontinued
BIIB078 (Biogen/Ionis) Phase 1: discontinued March 2022, no clinical benefit despite biomarker reduction. WVE-004 (Wave) FOCUS-C9 Phase 1b/2a: achieved 48–50% poly-GP reduction (P<0.0001) but no clinical benefit on ALSFRS-R — discontinued. ⚠️ Trap: these were Phase 1/1b trials, not powered for efficacy; the failure is still scientifically informative but should not be overstated.
- F10
Rosa Rademakers (Mayo Clinic) was the senior/corresponding author on the DeJesus-Hernandez 2011 paper; Mariely DeJesus-Hernandez was first author. Rademakers won the 2025 Breakthrough Prize in Life Sciences for this discovery.
DeJesus-Hernandez et al. 2011 — corresponding author: Rosa Rademakers, Department of Neuroscience, Mayo Clinic Jacksonville. Breakthrough Prize 2025 confirmed by AFTD and Breakthrough Prize announcement.
- —
DPR proteins (dipeptide repeats) from RAN translation described ~2013
Ash et al. 2013 and Mori et al. 2013 (both Science) first described DPR proteins from C9ORF72 repeats